Le cours de chimie de première année est une illustration de la place de la chimie dans la compréhension des sujets qui intéressent les ingénieurs AgroParisTech : aliments, environnement, santé, etc.
Ce cours aborde les questions suivantes :
- propriétés moléculaires
- réactivité : radicaux et oxydation
- réactivité : notions de chimie orbitalaire
- Trois cours à l'amphi : commentaire de ce site internet ;
- Après le deuxième cours, TD en salle informatique sur l'obtention de propriétés moléculaires ; lien à venir sur cette page pour un complément ;
- Après le troisième cours, deuxième TD en salle info : encore des propriétés moléculaires, de la réactivité...
- Examen de 45 minutes le 27 novembre 2023 : 09 h 30 - 10 h 15 ;
- QCM papier + question ouverte rapidement rédigée ;
- En principe, le 14 novembre 2023 à 17 h 00, amphi de révision A-TB, B, C, C2 : mieux vaut poser des questions avant !
Le site n'est pas conçu pour de trop petits écrans (téléphones...) et préfère Firefox ou la famille Chromium (Chromium, Chrome, Edge) comme navigateur.
Lorsqu'une molécule est représentée dans l'espace, la figure permet généralement de faire tourner ou de zoomer sur le représentation en plaçant le pointeur dans la figure et en utilisant les boutons de la souris.
Les atomes des molécules sont généralement représentée avec le code couleur CPK, les couleurs étant aussi appliquée jusqu'à la moitié des liaisons dans lesquelles sont engagés les atomes. Les molécules sont souvent représentées en mode « licorice », c'est-à-dire des tubes qui figurent par le squelette de leurs liaisons, comme la molécule de glucose ci-dessus. Les surfaces, quand il y en a, sont colorées selon un schéma indiqué en légende.
Les graphiques de type nuage de points, fonction mathématiques etc. sont zoomables, exportables... Il suffit de passer la souris un peu au dessus et à droite pour faire apparaître les icônes des fonction disponibles.
Propriétés moléculaires
Pour obtenir une vin effervescent comme le Champagne, le vin blanc « tranquille » est enfermé dans une bouteille avec du sucre pour qu'une nouvelle fermentation y génère un peu d'alcool, des arômes et surtout du dioxyde carbone. Une visite dans une atelier champenois montre que, avant cette mise en bouteille, le vin subit un traitement par le froid qui vise à faire précipiter les sels de l'acide tartrique que contient le vin blanc avant prise de mousse.
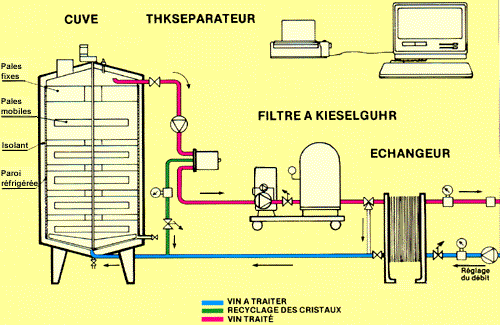
Autrement, on risque le « gerbage », c'est-à-dire l'expulsion d'une partie du champagne sous l'effet d'une effervesence soudaine et incontrôlée.
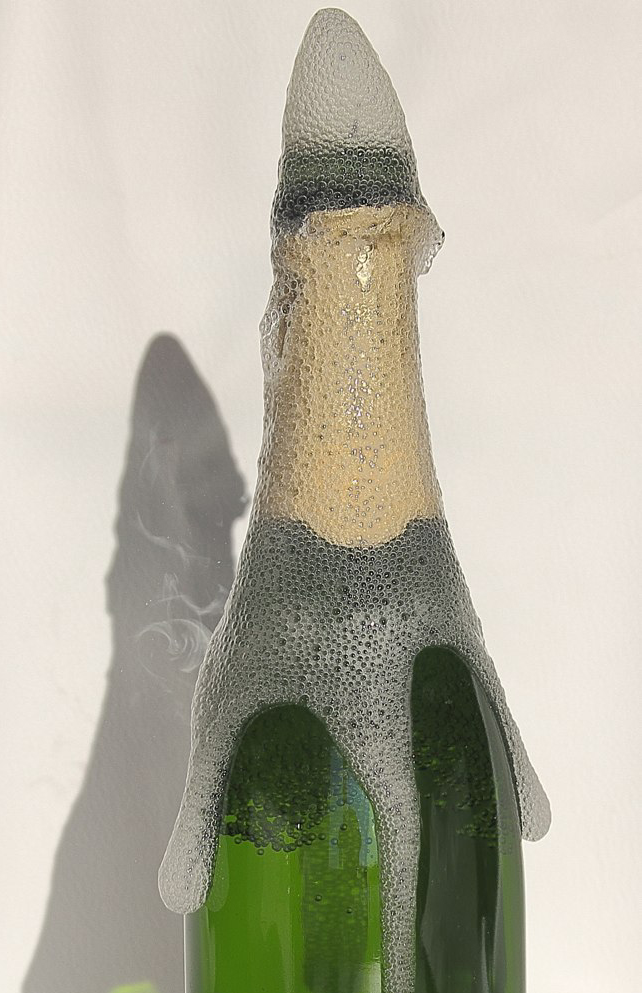
La littérature œnologique comprend plusieurs publications qui font état de la spécificité de l'expression du bouquet dans les vins effervescents :

Et on trouve aussi de nombreux articles sur la manière de laver, d'essuyer les verres, etc. pour obtenir une belle effervescence.

séché sans essuyage, et dans verre lavé normalement et essuyé avec un torchon.
À l'origine était la distribution des charges...
Dans cette partie, nous envisagerons des molécules sans qu'elles soient engagées dans des réactions, c'est-à-dire des échanges électroniques : formation ou rupture de liaison, gain ou perte d'électron. Les propriétés des molécules dérivent toutes d'interactions électrostatiques. Elles dépendent surtout de la répartition spatiale des charges, la matière étant globalement neutre.
Nous allons donc étudier les conséquences de l'existence des potentiels électrostatiques générés par les particules chargées que sont les noyaux et les électrons. Le potentiel électrostatique en un point est donné par l'expression classique :
$$E = \frac{1}{4 \pi \times \epsilon_0}\sum_{i}\frac{q_i}{r_i}$$où $q_i$ est la charge située à une distance $r_i$ de ce point
. Les charges positives sont portées par les noyaux ; elle sont- le rayon de l'atome d'oxygène est voisin de 60 pm ;
- le rayon du noyau d'oxygène est voisin de 30 fm ;
- longueur de la liaison O-O dans le peroxyde d'hydrogène : 145,7 pm
La figure suivante illustre cette probabilité pour la molécule de chlorure d'hydrogène. Les différentes couche colorées emboîtées correspondent à des isodensités électroniques. C'est au voisinage du noyau de chlore que la densité électronique est la plus grande ; elle correspond aux électrons de cœur de cet atome. Enfin, la somme de la densité électronique sur tout l'espace (supposé ne contenir qu'une molécule de chlorure d'hydrogène) est égale à $18 e$, puisque cette molécule contient au total $Z_{H} + Z_{Cl} = 18$ électrons.

La densité est maximale sur un anneau qui entoure l'atome de chlore.
Si les charges positives et négatives étaient parfaitement superposables, leurs effets électrostatiques s'annihileraient. L'exemple du chlorure d'hydrogène, sur une molécule pourtant simple, montre qu'il n'en est rien. C'est la répartition des électrons — position du barycentre, symétrie, éloignement moyen — par rapport aux noyaux qui confère plusieurs de leurs caractéristiques aux molécules :
charges partielles $\delta$ :
- au niveau d'un atome, somme de la charge nucléaire et des charges des électrons environnants ;
- généralement exprimée en unité élémentaire de charge $e$ : charge d'un proton ;
- c'est une grandeur fictive, car il n'est pas possible d'attribuer certainement une partie de la répartition spatiale d'électrons à un noyau particulier, sauf pour les ions monoatomiques. Ainsi, dans une molécule aussi simple que le chlorure d'hydrogène, comment définir au sein de la densité électronique, une frontière qui la partagerait, d'une part, en électrons « appartenant » à l'hydrogène et, d'autre part, en électrons « appartenant » au chlore ?
Il n'en reste pas moins que certaines régions au voisinage d'une molécule apparaissent excédentaires en charge positives et d'autres en charges négatives. Il est extrêmement commode d'attribuer, de manière plus ou moins empirique, une charge partielle $\delta$ pour décrire certaines propriétés des molécules. De trèsnombreuses méthodes par exemple : déterminer les charges qui, disposées à l'emplacement des noyaux, généreraient un potentiel électrostatique le plus proche possible de celui induit par les noyaux et la densité électronique spatiale existent pour ce faire, mais les exposer sortirait complètement du cadre de ce cours. En revanche, nous utiliserons en TD un logiciel qui permet de les évaluer et qui nous donnerait par exemple, pour la molécule de chlorure d'hydrogène gazeux : +0,15 $e$ sur l'hydrogène et -0,15 $e$ sur le chlore ; - La présence et la valeur des charges partielles dépendent :
- de l'électronégativité des atomes
- de la plasticité du nuage électronique. Cette dernière est elle même fonction de la taille de atomes, d'éventuels effets mésomères, etc.
moment dipolaire permanent $\mu$ :
- au niveau d'une liaison ou d'une molécule, lorsque les barycentres des charges positives et négatives ne sont pas confondus ;
- défini comme le produit de la charge par le vecteur qui les sépare (du - vers le +) ;
- exprimé en Debye : 1D = 3,33564 x 10-30 C m = 0,02081943 $e$ nm ;
- le moment dipolaire n'est pas une grandeur fictive ; il peut être mesuré ;
- le moment dipolaire moléculaire, donc la polarité, dépend :
- de l'électronégativité des atomes ;
- de la plasticité des liaisons : la liaison $\pi$ est plus déformable ("polarisable" : voir ci-après...) que la liaison $\sigma$ ;
- de la plasticité des nuages électroniques atomiques et donc du rayon atomique ;
- des distances entre centres positif et négatif.
polarisabilité $\alpha$ :
- facteur de proportionnalité entre la valeur $\vec{p}$ d'un dipôle induit et le champ $\vec{E}$ qui l'induit : $\vec{p} = \alpha \vec{E}$
- unité SI : C m2 V-1 ; on voit aussi dans la littérature une grandeur dérivée : $\alpha' = \frac{10^6}{4 \pi \epsilon_0 }\alpha$ homogène à un volume exprimé en cm3 ou, de manière plus commode, en Å3 (rappel : 1 Å = 10-10m)
- la polarisabilité n'est pas une grandeur fictive ; elle peut être (difficilement...) mesurée.
- la polarisabilité dépend :
- de la plasticité des liaisons : la liaison $\pi$ est plus polarisable que la liaison $\sigma$ ;
- de la plasticité des nuages électroniques atomiques et donc du rayon atomique ;
Charge partielle, moment dipolaire et polarisabilité ont pour conséquence l'existence de liaisons faibles qui confèrent aux molécules, en particulier condensées, de très nombreuses propriétés.
Petit rappel avant d'aller plus loin...
Il est utile de redonner les ordres de grandeur des énergies des liaisons qui ne sont pas qualifiées de « faibles ».
| Liaison | énergie kJ mol-1 | type |
|---|---|---|
| LiF | 1023 | ionique |
| MgO | 3900 | ionique |
| HH | 436 | covalente |
| CH | 415 | covalente |
| CC | 345 | covalente |
| C=C | 611 | covalente |
Liaisons faibles : forces liées aux charges partielles : énergies associées
Interaction entre un ion de charge $q_2$ et un dipôle $\mu_1$ distants de $r$
$$ E_{\text{ion-dipôle}} = -\frac{1}{r^2}\frac{ \mu_1 \times q_2}{4 \pi \times \varepsilon_r \times \varepsilon_0} $$$\varepsilon_r$ est la permittivité relative du milieu. Ordre de grandeur : 15 kJ mol-1.
Ces forces sont en jeu dans la solvatation des ions.
Liaison hydrogène
Elle peut être interprétée comme une interaction électrostatique entre la charge partielle négative d'un atome électronégatif et la charge partielle positive d'un hydrogène relié à un autre atome électronégatif (intra ou extra moléculaire). La liaison hydrogène est mieux décrite par la mécanique quantique, bien qu'elle soit assez éloignée d'une liaison covalente. On note cependant qu'elle possède une géométrie particulière : les trois atomes précédemment cités forment un angle compris dans une gamme déterminée, typiquement 160 à 180° pour OH-O dans la liaison entre deux molécules d'eau.
Ordre de grandeur : 8-40 kJ mol-1
| Liaison | énergie kJ mol-1 | molécules |
|---|---|---|
| O-H⋯N | 29 | eau-ammoniac |
| O-H⋯O | 21 | eau-eau |
| O-H⋯N | 8 | amide-eau |
| O-H⋯H | 18 | eau-oxonium |
Ces énergies expliquent en grande partie les propriétés physiques des espèces qui forment des liaisons hydrogène : point d'ébullition élevé par rapport à la masse moléculaire, viscosité, capacité thermique.
La liaison hydrogène participe à la stabilisation de la double hélice de l'ADN, des polymères comme la cellulose ou des structures secondaires des protéines. L'hélice $\alpha$, par exemple, est stabilisée par les liaisons hydrogène entre le N-H d'un acide aminé et le C=O de l'acide aminé situé quatre positions avant dans la chaîne peptidique.
Interactions dues aux dipôles : forces de van der Waals
Les forces de
- À la distance $r$ d'un dipôle, on observe un petit potentiel électrostatique en $\displaystyle \frac{1}{r}$
- À la distance $r$ d'un dipôle, on observe un petit potentiel électrostatique en $\displaystyle \frac{1}{r^2}$ qui résulte de la superposition de deux potentiels en $\displaystyle \frac{1}{r}$ de signes opposés et légèrement décalés ; à longue distance, ce potentiel décroit rapidement. Un dipôle exerce donc une force en $\displaystyle \frac{1}{r^3}$ sur une particule chargée.
- dans le champ en $\displaystyle \frac{1}{r^3}$ d'un dipôle, sur une paire de particules de charges opposées, c'est-à-dire sur un autre dipôle, apparaît une force qui résulte de la superposition de deux forces, légèrement décalées. Cette force est en $\displaystyle \frac{1}{r^7}$, elle dérive d'un potentiel en $\displaystyle \frac{1}{r^6}$ et constitue une force de van der Waals.
Suivant la nature des dipôles impliqués : permanents, induits ou instantanés, on distingue trois types de forces de van der Waals auxquelles sont associées trois expressions de l'énergie.
Forces de Keesom Willem Hendrik Keesom, 1876 - 1956, physicien néerlandais, spécialiste des cryotempératures
Elles s'exercent entre dipôles permanents : elles dépendent des moments dipolaires $\mu_1$ et $\mu_2$ des deux molécules entre lesquelles elles s'exercent. Elles dépendent aussi de l'alignement des deux dipôles l'un par rapport à l'autre et donc de l'agitation thermique du milieu. La présence du facteur $k_B \times T$ dénote l'effet de cette agitation.
$$ E_{\text{Keesom}} = -\frac{1}{r^6}\frac{\mu_1^2 \times \mu_2^2}{3(4 \pi \times \varepsilon_r \times \varepsilon_0)^2 \times k_B \times T} $$Ordre de grandeur : 2-20 kJ mol-1
Forces de Debye Peter Joseph Wilhelm Debye, 1884 - 1966, physicien et chimiste néerlando-américain, prix Nobel de chimie 1936. Ses contributions sont variées : diffraction X, conductivité, thermodynamique...
Ces forces apparaissent entre un dipôle et un dipôle induit. Leur formule est symétrique pour le rôle du moment dipolaire et de la polarisabilité :
$$ E_{\text{Debye}} = -\frac{1}{r^6}\frac{\mu_1^2 \times \alpha_2^2 + \mu_2^2 \times \alpha_1^2}{(4 \pi \times \varepsilon_r \times \varepsilon_0)^2} $$Ordre de grandeur : 2 kJ mol-1
Forces de LondonFritz London; 1900 - 1954, physicien américain d'origine allemande, spécialiste de mécanique quantique, professeur à l'université de Duke
ce sont des forces dites de dispersion, entre dipôles instantanés. La densité électronique est probabiliste : même sur une molécule très symétrique, même sur un atome isolé (a priori sphérique !!), il y a une forte chance à tout moment pour que celle-ci ne soit pas répartie à travers la molécule pour donner le même barycentre que les noyaux, ce qui crée un léger moment dipolaire. Le facteur $h\nu$ est relié au potentiel d'ionisation des molécules et c'est une sorte de descripteur, en plus des polarisabilités, de la plasticité de la distribution électronique
$$ E_{\text{London}} = -\frac{1}{r^6}\frac{3}{4}\frac{h\nu \alpha_1 \times \alpha_2}{(4 \pi \times \varepsilon_r \times \varepsilon_0)^2} $$Ordre de grandeur : 4-40 kJ mol-1
Modèle de Lennard-JonesJohn Edward Lennard-Jones, 1894-1954, chimiste et physicien théoricien britannique, professeur à Bristol et Cambridge. Il est considéré comme l'un des fondateur de la chimie numérique
Les forces de van der Waals sont globalement
$r$ est la distance entre deux atomes. Les paramètres du modèle sont :
- soit $r_m$, la distance moyenne de liaison, soit $\sigma=2^{-1/6} r_m$
- $\varepsilon$ l'énergie telle que $E_{LJ}(r_m) = -\varepsilon$
$\varepsilon$ est la notation consacrée par l'usage et n'a rien à voir avec la permittivité qui apparaît dans les expressions d'électrostatique ou de van der Waals.
$\pi$-stacking (interactions d'empilement aromatique)
Les systèmes aromatiques cycliques présentent une tendance à s'organiser sous l'effet d'interactions attractives non covalentes dues aux systèmes électroniques $\pi$. Il s'agit de la résultante de forces de dispersion et d'interactions

Le $\pi$-stacking contribue à stabiliser les hélices d'acide désoxyribonucléique par interaction entres bases adjacentes sur un même brin. Il est aussi impliqué dans la structurations tridimensionnelle des protéines ou dans le stabilisation des complexes. La figure qui suit montre ainsi le complexe formé entre l'acétylchloestérase humaine et le donépézil, médicament utilisé dans le traitement de la maladie d'Alzheimer.
Conséquences des liaisons faibles
Solubilité
La solubilité est la quantité volumique de matière dissoute dans une solution en équilibre avec une phase pure c'est-à-dire, si on considère une espèce $\ce{C}$, l'avancement volumique dans un solvant $\ce{S}$ de la réaction :
$\qquad \ce{C_{pur} <=> C_{en \, solution}} \qquad K$
Dans le solvant pur et sans autre réaction : $\ce{[C_{en \, solution}] = K\times C^{0}}$
$\qquad K = \exp(-\Delta G^0_{sol}/RT) \qquad \qquad \Delta G^0_{sol} = \Delta H^0_{sol} - T \Delta S^0_{sol}$
Cet avancement dépend de l'enthalpie libre de réaction et, par conséquent, de termes enthalpiques et entropiques. Leur bilan dépend du soluté et du solvant.
Composées ioniques dans l'eau
La présence des charges est déterminantes, puisque les forces coulombiennes dominent. Le bilan enthalpique comprend :
- un terme positif : énergie réticulaire $E_{ret}$, rupture du réseau cristallin (fortes liaisons électrostatiques)
- un terme négatif : l'enthalpie d'hydratation des ions libérés
- bilan positif : exemple $\ce{NaCl}$, la solubilité augmente au chauffage
- bilan négatif : exemple $\ce{CaCO3}$, la solubilité diminue au chauffage (entartrage, dissolution des calcaires océaniques profonds)
Le bilan entropique comprend :
- un terme positif de perte de l'ordre du réseau cristallin
- un terme positif de perte très partielle de la structuration intermoléculaire de l'eau par les liaisons hydrogène
- un terme négatif de structuration de sphères d'hydratation. Elle peut être faible autour de Ag+, forte autour de Na+ voire très forte comme autour de Ca2+
| composé | $E_{ret}$ kJ mol-1 |
$n_{\ce{H2O}}$ cation |
$n_{\ce{H2O}}$ anion |
enthalpie kJ mol-1 |
entropie J mol-1 K-1 |
|---|---|---|---|---|---|
| $\ce{NaCl}$ | 771 | 3,5 | 2 | 3,8 | 43 |
| $\ce{AgCl}$ | 916 | 3,1 | 2 | 65 | 33 |
| $\ce{CaCO3}$ | 2814 | 7,2 | 4 | -12 | -200 |
Composés hydrophobes dans l'eau
Le cas des composés hydrophobes est intéressant. Leur faible solubilité est souvent attribuée à l'absence de liaison hydrogène avec l'eau, mais cette analyse est loin d'être suffisante. En effet, le bilan enthalpique reste faible : des liaisons de Van der Waals sont rompues dans la masse du composé hydrophobe, des liaisons hydrogène dans la masse de l'eau, mais des liaisons de van der Waals s'établissent entre le composé hydrophobe et l'eau. Il peut être légèrement positif.
Plus intéressant que celui des composés ioniques, le bilan entropique comprend :
- un terme positif de déstructuration du liquide du composé hydrophobe pur (liquide ou solide) ; il est très faible, car ce type de liquide est peu structuré, même quand il est le siège de liaisons de van der Waals
- un terme positif de perte très partielle de la structuration intermoléculaire de l'eau par les liaisons hydrogène
- un terme important et toujours négatif de structuration de l'eau en cages de solvant autour des molécules de composés hydrophobe comme sur la figure suivante :
Ce bilan, généralement négatif, explique la faible solubilité des composés hydrophobes et se traduit par une enthalpie libre standard de dissolution souvent positive :
$$ \Delta G^0_{sol} = \Delta H^0_{sol}(moyen) - T \Delta S^0_{sol}(négatif) \qquad \Delta G^0_{sol} > 0 $$Le bilan enthaplique est moyen, mais non nul : la rupture de nombreuses interactions de van der Waals dans le composé hydrophobe pur est partiellement compensée par le même genre de liaisons avec l'eau ; l'eau perd elle-même des liaisons faibles. La solubilité des alcanes, par exemple, augmente avec la température.
Coefficient de partage octanol/eau - logP
Que ce soit en matière d'environnement, de biologie ou d'agroalimentaire, il est très utile de savoir comment des composés organiques vont se distribuer, c'est-à-dire dans quelle phase ils vont avoir tendance à se situer, lorsque plusieurs compartiments existent : aqueux (eau, plasma, etc.) ou hydrophobe (membrane, tissus gras, huile, etc.)
Pour ce faire, on s'intéresse au coefficient de partage des composés entre l'eau et l'octanol :
Soit un composé $\textbf{C}$ susceptible de se dissoudre dans l'octanol $\textbf{o}$ et dans l'eau $\textbf{w}$. Lorsque l'équilibre est atteint, le potentiel chimique de $\textbf{C}$ est égal dans $\textbf{o}$ et $\textbf{w}$ :
$$ \mu_{\textbf{C}}^{\textbf{o}} = \mu_{\textbf{C}}^{\circ \textbf{o}}(T) + RT \ln\frac{[\textbf{C}]^{\textbf{o}}}{C^\circ} = \mu_{\textbf{C}}^{\circ \textbf{w}}(T) + RT \ln\frac{[\textbf{C}]^{\textbf{w}}}{C^\circ} = \mu_{\textbf{C}}^{\textbf{w}}$$Il en découle :
$$ P = K_{ow} = \frac{[\textbf{C}]^{\textbf{o}}}{[\textbf{C}]^{\textbf{w}}} = \exp\left[\frac{\mu_{\textbf{C}}^{\circ \textbf{w}}(T) -\mu_{\textbf{C}}^{\circ \textbf{o}}(T)}{RT} \right] $$Le coefficient de partage octanol/eau est une mesure usuelle de l'hydrophobie ou de la lipophilie d'une molécule. C'est le logarithme du coefficient partage qui est la grandeur la plus employée :
$$ \log P = \log K_{ow}$$L'utilisation de l'octanol comme solvant organique permet d'avoir une échelle plus nuancée pour les lipophilies intermédiaires que ne le serait l'échelle établie pour le partage alcane/eau. La figure ci-dessous donne la valeur de $\log P$ pour trois composés de polarité et de tailles différentes.

Le coefficient de partage octanol/eau est une grandeur fondamentale pour comprendre la localisation et le transport de nombreuses espèces chimiques. Cette grandeur est d'un emploi très large : deux exemples sont donnés dans le lignes qui suivent.
Facteur de bioconcentration
Le « facteur de bioconcentration » (FBC ou BCF en anglais) désigne le rapport entre la concentration d'un composé chimique un organisme vivant et la concentration dans le milieu. C'est une grandeur très importante pour caractériser l'accumulation des polluants dans les êtres vivants (bioaccumulation) le long des chaînes trophiques telle que la schématise la figure suivante.

Pour les polluants organiques, le facteur de bioconcentration est lié à la lipophilie puisque de nombreuses molécules organiques sont solubles dans les solvants organiques.
Adsorption de polluants organiques dans le sol
Les hydrocarbures aromatiques polycycliques sont des polluants très toxiques. De par leur géométrie plane et leurs nombreux nuages $\pi$, ils s'adsorbent sur les particules de la phase solide du sol. Cette adsorption apparaît liée à l'hydrophobicité.
Effet d'autres constituants sur $\log P$
Lorsque l'un des solvants (octanol ou eau) n'est pas pur, la valeur du potentiel standard de $\textbf{C}$ dans ce solvant n'est plus égale à $\mu_{\textbf{C}}^{\circ \textbf{o}}$ ou $\mu_{\textbf{C}}^{\circ \textbf{w}}$. La valeur de $K_{ow}$ peut s'en trouver modifiée.
Cet effet est par exemple mis à profit dans l'utilisation de sel pour améliorer le relargage lors d'une extraction liquide-liquide (voir TP d'orga...). La modification des valeurs de $K_{ow}$ permet aussi de comprendre l'influence sur le comportement des molécules de la salinité de milieux comme l'eau de mer ou le plasma sanguin.
Facteur de distribution - logD
Le facteur de distribution $D$ est la version dépendante du pH, et donc de l'ionisation, du coefficient de partage. La définition de $K_{ow} = P$ étant, pour une espèce $\textbf{C}$ :
$$ K_{ow} = \frac{[\textbf{C}]^{\textbf{o}} }{[\textbf{C}]^{\textbf{w}} } $$celle de $D$ est :
$$ D = \frac{[\textbf{C}]^{\textbf{o}} + \sum [\textbf{C}_\text{ionisé}]^{\textbf{o}}}{[\textbf{C}]^{\textbf{w}} + \sum [\textbf{C}_\text{ionisé}]^{\textbf{w}}} $$En pratique, il est rare que les molécules soient ionisées dans la phase organique ; l'expression de $D$ se simplifie alors :
$$ D = \frac{[\textbf{C}]^{\textbf{o}}}{[\textbf{C}]^{\textbf{w}} \left[ % 1 + \sum \frac{[\textbf{C}_{\text{ionisé}}]^{\textbf{w}}} {[\textbf{C}]^{\textbf{w}}} % \right]} = \frac{K_{ow}} {1 + \sum \frac{[\textbf{C}_{\text{ionisé}}]^{\textbf{w}}} {[\textbf{C}]^{\textbf{w}}}} $$$\displaystyle \sum \frac{[\textbf{C}_{\text{ionisé}}]^{\textbf{w}}} {[\textbf{C}]^{\textbf{w}}}$ est une fonction du pH, et des constantes d'acidité.
Pratiquement, les acides carboxyliques sont sous forme moléculaire en dessous de leur pKa et sous forme ionisée de carboxylate au dessus. Au contraire, les amines sont protonées sous forme d'ammonium aux pH inférieurs au pKa de leur acide conjugué. Les composés amphotères manifestent un comportement plus compliqué. La figure suivante rend compte de ces différences.
acide octanoïque (bleu), 1-aminooctane (orange), acide 8-aminooctanoïque (vert)
Le $\log D$, pour les molécules acido-basiques, est beaucoup plus explicatif du comportement des molécules que ne l'est le $\log P$. Le $\log D$ fait par exemple partie des critères de la
- moins de 5 hydrogènes donneurs de liaisons hydrogènes
- moins de 10 atomes accepteurs de liaison hydrogène
- masse moléculaire inférieure à 500 Daltons
- log D inférieur à 5
La figure suivante montre comment par exemple le $\log D$ permet de modéliser les propriétés d'une enzyme :
le KM peut se modéliser en fonction du volume molaire (encombrement) et du $\log D$ mieux que du $\log P$
Constante de Henry
La loi de
Lorsque l'équilibre est atteint, le potentiel chimique de $\textbf{G}$ est égal dans $\textbf{L}$ et $\textbf{A}$ :
$$ \mu_{\textbf{G}}^{\textbf{L}} = \mu_{\textbf{G}}^{\circ \textbf{L}}(T) + RT \ln\frac{[\textbf{G}]^{\textbf{L}}}{C^\circ} = \mu_{\textbf{G}}^{\circ \textbf{A}}(T) + RT \ln\frac{P_{\textbf{G}}^{\textbf{A}}}{P^\circ} = \mu_{\textbf{G}}^{\textbf{A}} $$et il en découle :
$$ K_H = \frac{[\textbf{G}]^{\textbf{L}}\times P^\circ}{P_{\textbf{G}}^{\textbf{A}}\times C^\circ} = \exp\left[\frac{\mu_{\textbf{G}}^{\circ \textbf{A}}(T) - \mu_{\textbf{G}}^{\circ \textbf{L}}(T)}{RT} \right]$$$K_H$ est la constante thermodynamique de l'équilibre. La loi de Henry énonce alors : « À température constante et à l'équilibre, la quantité de gaz dissous dans un liquide est proportionnelle à la pression partielle qu'exerce ce gaz sur le liquide. »
Remarque importante : la loi de Henry est très souvent mise en œuvre sans appliquer les conventions de la thermodynamique chimique. Ceci ne remet pas en cause le principe de proportionnalité, mais peut introduire des unités pour la constante de Henry, lorsque les activités dans la solution et/ou dans la phase gazeuse ne sont pas rapportées aux activités de référence $C^\circ$ et $P^\circ$.
Dissolutions des gaz dans l'eau de mer
La loi de Henry est par exemple utilisée pour prévoir la répartition du dioxyde de carbone entre l'atmosphère terrestre et les océans. La modélisation est assez complexe :
- le dioxyde de carbone moléculaire est aussi en équilibre, mais seulement dans l'océan, avec ses formes dissoutes bases conjuguées
- la constante de Henry varie avec la température et l'océan présente un profil de température vertical marqué.
Les résultats en sont résumés dans la figure qui suit.

Noter que l'augmentation de la teneur en dioxyde de carbone ne s'accompagne pas d'une augmentation de celle en carbonates dissous (ce sont des bases), mais de celle en hydrogénocarbonates (acides conjugués des carbonates) : l'océan s'acidifie.
Comme le $\log P$, la constante de Henry est modifiée par la présence d'autres constituants que le solvant et le gaz qui s'y dissout. La figure suivante montre comment la solubilité du dioxygène diminue lorsqu'un sel est dissous dans l'eau. La pression partielle de dioxygène étant constante dans l'atmosphère (à une altitude donnée, bien évidemment), la variation de la solubilité correspond à une variation de la constante de Henry.
Libération des arômes par les aliments
Les aromaticiens utilisent aussi beaucoup les constantes de Henry pour prévoir comment les molécules odorantes présentes dans un aliment vont passer dans l'atmosphère au dessus de l'aliment et seront perçues par voie orthonasale (par le nez, en humant l'aliment) ou dans l'atmosphère de la cavité buccale et seront perçues par voir rétronasale (par l'arrière du palais pour atteindre les fosses nasales, en ingérant et mastiquant l'aliment). En effet, les constituants des arômes manifestent des comportement très variés vis-à-vis des deux solvants principaux que sont les phases aqueuses et lipidiques des aliments. Le tableau suivant donne les constantes de Henry (affligées d'une unité) des quelques constituants de l'arôme de café. À concentration constante dans la solution aqueuse : le café. la perception d'une molécule d'arôme dépend de la constante de Henry et de la sensibilité de l'épithélium olfactif à cette molécule.
| Composé | formule | KH x 10-4 Pa |
|---|---|---|
| 2,3-butanedione |  |
10,0 |
| 2,3-pentanedione |  |
38,8 |
| 3-méthybutanal |  |
89,3 |
| benzaldéhyde |  |
13,8 |
| acétaldéhyde |  |
47,1 |
| furfural |  |
1,8 |
| 2,5-diméthylpyrazine |  |
1,9 |
| 5-méthylfurfural |  |
8,3 |
Une autre conséquence physique : la tension superficielle
À l'intérieur d'un liquide, chaque molécule entretient des interactions faibles : liaisons hydrogène, interactions de van der Waals, avec ses voisines et l'ensemble des molécules qui constituent le liquide trouve ainsi une configuration d'énergie minimale et stable. Il faudra par exemple apporter de l'énergie, la chaleur latente de vaporisation, pour rompre les liaisons faibles internes, éloigner les molécules les unes des autres et vaporiser ainsi le liquide.
Ces liaisons faibles expliquent aussi le phénomène de tension superficielle : à l'interface avec un autre fluide, les molécules d'un liquide forment un nombre limité de liaisons faibles avec ce fluide. Si ces liaisons étaient aussi nombreuses et fortes qu'au sein du liquide lui-même, il se mélangerait avec le fluide et il n'y aurait pas d'interface : c'est ce qu'on observe par exemple entre l'eau et l'éthanol qui sont miscibles en toutes proportions.

La présence d'une interface réduit ainsi le nombre d'interactions stabilisatrices et entraîne donc un surcoût énergétique surfacique : c'est la tension superficielle. Pour limiter ce coût, le liquide cherche à adopter une forme, généralement sphérique, qui minimise l'aire de l'interface : c'est ce que fait le whisky du capitaine Haddock lorsqu'il n'est plus maintenu au fond de son verre par la gravitation.
À l'inverse, il faut de l'énergie pour augmenter la taille de l'interface. Le travail $\delta W$ nécessaire pour accroître de $dA$ la surface $A$ d'une interface est donné par :
$\delta W = \gamma dA$
où $\gamma$ est en pratique dénommé « tension superficielle ».
À une toute autre échelle (environ 5 x 1020 fois moins de molécules !!), le même phénomène est illustré sur la figure suivante qui montre comment une lame d'eau parallélépipédique placée dans le vide (et en l'absence de gravité) se transforme spontanément en goutte sphérique. Il faut noter que, du fait d'une température non nulle, une partie de l'eau s'évapore (seulement quelques molécules) pour former un gaz à la pression de vapeur saturante. Ne pas hésiter à utiliser la souris pour faire tourner la figure (éventuellement zoomer) pour voir la lame d'eau sous le meilleur angle possible.
L'animation tourne en boucle.
L'exemple des gouttes met en jeu une interface liquide/gaz. L'usage a consacré le terme de tension interfaciale pour les interfaces entre deux milieux denses, mais le terme « tension superficielle » est correct dans tous les cas.
Le tableau ci dessous reprend quelques valeurs de tension superficielle dans les interface avec l'air :
| liquide | température (°C) | $\gamma$ mN m-1 |
|---|---|---|
| acide acétique 10% | 30 | 54,6 |
| eau | 20 | 72,8 |
| eau | 37 | 70 |
| eau | 100 | 58,9 |
| éthanol 11% | 25 | 46,0 |
| éthanol | 20 | 22,2 |
| glycérol | 20 | 63 |
| huile | 20 | 20 |
| octane | 20 | 21,8 |
| mercure | 20 | 436 |
| plasma sanguin | 37 | 73 |
| sirop (saccharose 55 %) | 20 | 76,5 |
| surfactant pulmonaire + eau | 37 | 25 |
La tension superficielle résulte des liaisons faibles : elle est donc plus élevée chez les espèces sièges de liaisons hydrogène que ches les alcanes et croît avec le nombre d'atomes, en raison des forces de van der Waals.
La tension superficielle intervient dans de nombreux systèmes. On peut citer par exemple :
L'instabilité des émulsions
Pour un volume donné de liquide (dans le tableau ci-dessous, il s'agit du volume $V$ d'une sphère de rayon $r$) mis sous forme de gouttes, la surface est d'autant est plus grande que le nombre de goutte est élevé : la surface de deux gouttes sphériques de volume $V/2$ est supérieure à celle d'une goutte sphérique de volume $V$, etc. :
| nombre de gouttes | volume total | volume d'une goutte | rayon d'une goutte | surface totale |
|---|---|---|---|---|
| $1$ | $\frac{4}{3}\pi \times r^3$ | $\frac{4}{3}\pi \times r^3$ | $r$ | $4 \times \pi \times r^2$ |
| $2$ | $\frac{4}{3}\pi \times r^3$ | $\frac{4}{3 \times 2}\pi \times r^3$ | $\frac{1}{2^{1/3}}r$ | $4 \times \pi \times r^2 \times 2^{1/3}$ |
| $N$ | $\frac{4}{3}\pi \times r^3$ | $\frac{4}{3 \times N}\pi \times r^3$ | $\frac{1}{N^{1/3}}r$ | $4 \times \pi \times r^2 \times N^{1/3}$ |
Afin de minimiser l'énergie interfaciale et donc la surface, les gouttes d'une émulsion (par exemple d'huile dans l'eau comme la mayonnaise) ont donc tendance à fusionner dès qu'elles rentrent en contact. Une fois formée (ou pendant leur formation) les émulsions doivent donc être stabilisées en empêchant le mouvement des gouttes par l'emploi d'épaississants ou en diminuant la tension superficielle grâce à des émulsifiants qui stabilisent l'interface.

Ci-dessus est représentée une molécule d'une phosphatidylcholine. C'est un bon exemple d'émulsifiant et un des constituants des lécithines couramment employées en agroalimentaire. À la frontière entre de l'eau et de l'huile, les deux chaînes aliphatiques forment des liaisons de van de Waals avec les molécules de triglycérides, tandis que les groupements phosphate et ammonium sont solvatés par l'eau. Une molécule phosphatidylcholine établit ainsi des liaisons faibles avec l'eau et avec l'huile, stabilisant ainsi l'interface entre les deux liquides.
En agroalimentaire ou en cosmétique, les molécules de tensio-actifs ou émulsifiants se voient attribuer un indice HLB pour les caractériser. Des exemples seront donnés en TD.
l'égouttage des produits
La viscosité et la gravité (ou la force centrifuge) sont les facteurs déterminants de l'écoulement donc de l'égouttage des produits mais, lorsque la quantité de liquide diminue, ce n'est plus une veine continue qui s'écoule et des gouttes doivent être formées. Cette formation est plus difficile lorsque la tension superficielle est forte. La figure suivante montre le processus d'égouttage d'une frite qui sort de l'huile chaude : en plus d'être tirée par son poids, l'huile est poussée par la bulle de la vapeur s'échappe qui s'échappe de la frite (la pomme de terre contient plus de 75% d'eau). Lorsque le poids et la force de la pression de la vapeur sont supérieurs à la tension superficielle, la goutte se détache.

Photos tirées de la thèse de Maxime Touffet (AgroParisTech, 2018) :
Transferts et réactivité de l’huile au cours du procédé de friture
Les vapeurs sursaturantes
Nous envisagerons le cas de la formation des gouttelettes de pluie à partir de la vapeur d'eau. Elle doit commencer par la condensation de gouttelettes microscopiques, de rayon très faible. Or pour, une goutte sphérique, le rapport surface sur volume est inversement proportionnel au rayon. Il en va de même pour le bilan de la condensation : le coût de la construction de l'interface eau/air est proportionnel à la surface, tandis que la chaleur de condensation est proportionnelle au volume de la goutte.
Une modélisation simplifiée permet de déterminer qu'il faut une pression de vapeur d'eau égale à au moins trois fois la pression de vapeur saturante pour que les gouttelettes se forment spontanément (consultable, mais en dehors de ce cours : microphysique des nuages.) La tension superficielle empêche donc qu'il pleuve !
En fait, des poussières, des microcristaux issus des embruns et présents dans l'atmosphère absorbent de l'eau et les gouttelettes ainsi formées sont assez grosses et leur contenu a une tension superficielle assez faible pour que le processus de condensation puisse effectivement avoir lieu.
De même, le formation de microbulles est-elle peu favorisée dans une boisson carbonatée limpide, de sorte qu'elle peut contenir, même à l'air libre une quantité de dioxyde carbone dissous qui correspondrait à une pression de CO2 de plusieurs bars. L'introduction de bonbons dans un soda peut conduire à des dégazages brutaux, comme dans la très connue « Mentos eruption » :
Une explication plus détaillée est disponible ici
On trouve là des explications sur ce qui a été décrit au sujet du champagne :
- les microcristaux des sels de l'acide tartrique peuvent être des sites de formation des bulles et provoquer le gerbage : il faut les éliminer ;
- les fibres de celluloses laissées par l'essuyage au torchon sont creuses et contiennent des microbulles d'air qui sont des noyaux de formation des bulles de dioxyde de carbone ;
- les bulles augmentent le volume de gaz en contact avec le liquide, se chargent de composés volatils : loi de Henry et les transportent à la surface du liquide, dévoilant ainsi l'arôme du Champagne.
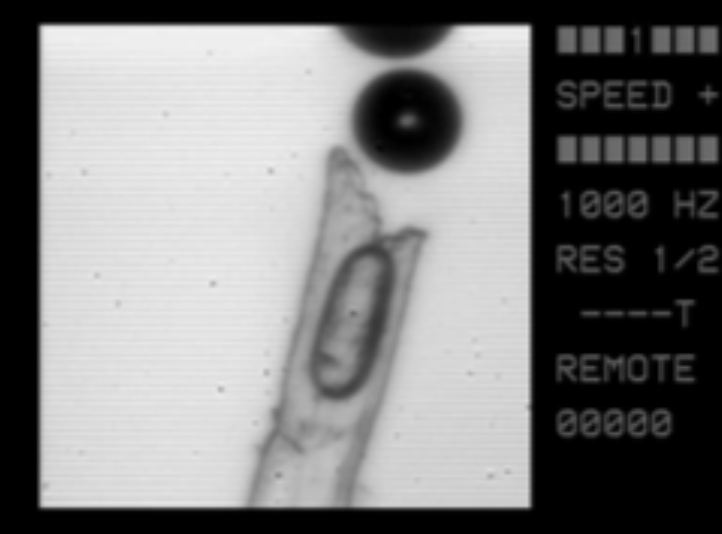
Diamètre de la fibre : environ 40 µm ; cliché G. Liger-Belair Université de Reims.
L'angle de contact
Définition
C'est l'angle de contact d'un liquide sur un solide qui est l'angle $\theta$ formé par la surface du solide et celle du liquide le long de leur ligne de contact :

Rôle des liaisons faibles
Il dépend notamment de la tension superficielle de l'interface liquide-gaz et de l'interface liquide-solide. Il mesure ainsi la mouillabilité d'une surface par un liquide qui dépend donc in fine de liaisons faibles. Les deux figures suivantes illustrent comment les liaisons hydrogènes entre un solide et l'eau peuvent rendre une surface mouillable.
L'animation tourne en boucle et n'est pas forcément bien orientée au chargement de la page.
L'animation tourne en boucle et n'est pas forcément bien orientée au chargement de la page.
Quelques valeurs
Les valeurs de l'angle de contact sont recouvrent bien la mouillabilité des matériaux par les liquides, ainsi que l'illustrent les valeurs données ci-après en exemple. Ainsi, l'eau mouille le verre parfaitement dégraissé, mais pas le Téflon (marque déposée du polytétrafluoroéthylène).
| liquide | surface solide | gaz | angle |
|---|---|---|---|
| eau | Téflon | air | 108° |
| glycérol | Téflon | air | 100° |
| $\ce{CCl4}$ | Téflon | air | 36° |
| eau | inox 316 | air | 71° |
| eau | cuivre | air | 86° |
| éthanol | cuivre | air | 14−19° |
| eau | verre | air | ≈ 0° |
| mercure | verre | air | ≈ 130° |
Comme la tension superficielle, l'angle de contact varie avec la température.
Un autre exemple à propos de la tension superficielle : l'effet fakir
Les propriétés des liaisons faibles et particulier de leur conséquences sur les tensions interfaciales jouent donc un rôle majeur dans l'environnement, en biologie, etc. Pour conclure, voici à titre d'illustration les deux clés de ce qu'on appelle l'effet lotus, c'est-à-dire le caractère superhydrophobe de certaines cuticules foliaires, comme celles du lotus, qui laissent véritablement glisser l'eau.

Un examen microscopique montre que la cuticule des feuilles de lotus est hérissée de cônes de cires cristallisée.

Une goutte d'eau à la surface d'une feuille de lotus se comporte alors ainsi :

Les gouttes d'eau sont comme posées sur les pointes que constituent les cônes de cire. La surface de contact entre l'eau et la cire est particulièrement faible. En effet, au niveau des pointes des cônes, l'angle de contact θ est très grand ; par ailleurs, la tension superficielle de l'interface eau/air empêche l'eau de descendre entre les cônes de cire. Le contact de limite donc à l'extrémité des cônes. On parle alors d'effet fakir ; cet effet confère aux feuilles de lotus, à l'échelle macroscopique, le caractère superhydrophobe qui permet à l'eau de glisser sur elles.
Systèmes hors d'équilibre
Systèmes hors d'équilibre : le cas des réactions impliquant l'oxygène
Introduction
Dans la première partie du cours, il a été question de propriétés des molécules, sans envisager de réaction, c'est-à-dire de mouvement d'électrons. Ces mouvements peuvent être de simples échanges : gain ou perte, lors de réactions d'oxydoréduction, soit des réorganisations dans les orbitales moléculaires, lorsque des liaisons sont rompues ou créées. Ces mouvements conduisent statistiquement et au bout d'un certain temps, à une organisation stable des cortèges électroniques, autrement dit à la formation des produits thermodynamiquement les plus stables.
Cependant, nombre de systèmes qui nous intéressent : atmosphère, cellule ou organisme vivant, etc. ne sont pas fermés et échangent de l'énergie ou de la matière avec l'extérieur. Ce n'est alors pas toujours l'équilibre thermodynamique qui gouverne leur composition : ce peut être la vitesse à laquelle sont consommées ou produites les espèces chimiques qui les composent.
Nous envisagerons ici plusieurs exemples dans lesquelles c'est effectivement la vitesse qui doit être étudiée pour comprendre leur fonctionnement. Dans tous ces exemples, l'oxygène joue un rôle important.
L'ozone stratosphérique
L'ozone $\ce{O_3}$ est naturellement présente dans la stratosphère : à 22,5 km d'altitude, une pression partielle $P_{\ce{O3}}$ d'environ 29 mPa est mesurée. La présence de ce gaz est d'une importance vitale pour la planète. En effet, l'ozone absorbe les ultraviolets solaires entre 200 et 300 nm et protège la biosphère de ce rayonnement destructeur. Ce n'est pas la seule abondance du dioxygène qui peut explique la présence de l'ozone. On pourrait imaginer un équilibre $\ce{3 O2 <=> 2 O3}$, mais sa constante thermodynamique de est très petite. Les donnée ci-dessous permettent de la calculer à T = 214 K, température moyenne à cette altitude.
| molécule | $\Delta H^0_f$ (kJ mol-1) | $\Delta S^0_f$ (J mol-1 K-1) |
|---|---|---|
| $\ce{O2}$ | 0 | 205,4 |
| $\ce{O3}$ | 142,76 | 238,9 |
On obtient K= 1,2 10-77 (détail du calcul avec ce lien) de sorte que la pression partielle d'ozone à l'équilibre devrait tendre vers zéro. Le calcul donne $P_{\ce{O3}}$ = 2,510-37 Pa.
Modèle de ChapmanSydney Chapman, astronome et géophysicien britannique, (29 janvier 1888, Eccles - 19 juin 1970, Boulder, Colorado). A travaillé principalement sur la dynamique des gaz, le magnétisme terrestre et interplanétaire, et l'ionosphère. Auteur de Sydney Chapman, "A Theory of Upper-Atmospheric Ozone," Memoirs of the Royal Meteorological Society 3 (26),103-25 (1930).
En fait, la stratosphère ne constitue pas un système thermodynamique isolé à l'équilibre : elle est notamment traversée par un
- $\ce{O2 + hν -> 2 O}$ constante de vitesse $k_1$ en s-1
- $\ce{O + O2 + M -> O3 + M}$ constante de vitesse $k_2$ en cm6 mol-2 s-1
- $\ce{O3 + hν -> O + O2}$ constante de vitesse $k_3$ en s-1
- $\ce{O + O3 -> 2 O2}$ constante de vitesse $k_4$ en cm3 mol-1 s-1
En appliquant l'approximation des états quasi stationnaires aux composés minoritaires : $\ce{O}$ et $\ce{O3}$ et en considérant que, du fait qu'elle implique deux composés minoritaires, la vitesse du quatrième acte est sans doute petite devant les autres,
Les constantes $k_2$ et $k_4$ dépendent de la température selon la loi d'
Ce modèle surestime la teneur réelle en ozone, mais décrit bien une « couche » à une altitude conforme aux observations. Pour avoir une estimation correcte de la pression réelle d'ozone, il faut a minima prendre en compte sa destruction provoquée par les oxydes d'azote et les oxydes d'hydrogène et décrite par au moins quatre actes élémentaires supplémentaires. La présence d'autres espèces d'origine anthropique, par exemple des chlorofluoroalcanes ou CFC, peut même provoquer une réduction inquiétante de la teneur en ozone mesurée et représente un réel danger pour la survie de la planète moins protégée contre les ultraviolets.
Oxydation de la matière organique
Le dioxygène est un oxydant puissant : $E^0_{[\ce{O2/H2O}]} = 1{,}23V$ et son activité dans la biosphère est élevée : $a_{\ce{O2}} = 0{,}2$. Par ailleurs, la matière organique est oxydable. On peut citer par exemple : $E^0_{[\ce{CO2/CH4}]} = 0{,}17 V$, $E^0_{[\ce{CO2/C}]} = 0{,}21 V$ ou encore : $E^0_{[\ce{CO2/HCOOH}]} = -0{,}11 V$. Le dioxyde de carbone devrait par conséquent être la principale espèce du carbone dans la lithosphère. Le carbone zéro est pourtant abondant et très stable : un morceau de paraffine peut rester à l'air, isolé ou nom,
« Given the exothermicity of these reactions, it is surprising that $\ce{O2}$ can exist in the atmosphere in presence of organic compounds ».
L'oxydation est pourtant possible, et à la différence de la couche d'ozone, un état stationnaire n'est alors pas atteint : par certains processus comme les combustions ou la respiration, la matière organique peut être complètement oxydée comme le prévoit la thermodynamique, mais à une vitesse qui dépend du processus.
Pour comprendre la réactivité particulière du dioxygène, il est nécessaire d'examiner l'organisation de cette molécule.
La structure électronique du dioxygène
Pour savoir où se situent les électrons — les moteurs des réactions — dans une molécule, il faut déterminer les orbitales moléculaires : les lieux de l'espace où les électrons d'une énergie, par ailleurs quantifiée, ont une probabilité de se situer. Ces orbitales moléculaires sont les équivalents des orbitales atomiques, mais pour un système à plusieurs noyaux. Elles sont impossible à déterminer par unCombinaison linéaire d'orbitales atomiques
Cas du dihydrogène
Commençons par examiner le cas simple du dihydrogène. Cette molécule de dihydrogène possède, dans la représentation de
- les électrons de cœur restent principalement sous l'influence du noyau de leur atome d'origine ;
- les orbitales de électrons de valence sont des combinaisons linéaires des orbitales des électrons de valence des atomes de la molécule.
Dans le cas des atomes d'hydrogène, il n'y a qu'un électron par atome, c'est un électron de valence et il occupe, dans un atome d'hydrogène seul, l'orbitale $1s$ . Soit alors deux atomes d'hydrogène et les orbitales qui correspondent à leur état fondamental d'énergie $E_{\ce{H}}$ : $\psi_1$ l'orbitale $1s$ d'un des atomes et $\psi_2$ l'orbitale $1s$ de l'autre. L'hypothèse CLAO est que les orbitales moléculaires $\phi$ du dihydrogène prennent la forme :
$$ \phi=c_1 \psi_1 + c_2 \psi_2 $$Le calcul montre alors que
et
$$ \phi_{\sigma^*} = \frac{\psi_1-\psi_2}{\sqrt{2(1-S)}} $$où :
$$ S= \int \psi_2\,\psi_1^{*}\,\mathrm{d}V = \int \psi_2^{*}\,\psi_1\,\mathrm{d}V $$est appelée intégrale de recouvrement de $\psi_1$ et $\psi_2.$ Ces deux orbitales moléculaires, respectivement nommées $\sigma$ et $\sigma^*$, présentent des énergies $E_{\sigma}$ et $E_{\sigma^*}$ telles que :
$$ E_{\sigma} < E_{\ce{H}} < E_{\sigma^*} $$ $$ E_{\ce{H}} - E_{\sigma} < E_{\sigma^*} - E_{\ce{H}} $$ En raison de ce classement, les deux électrons du dihydrogène occupent, dans l'état fondamental, l'orbitale $\sigma$ ; les règles de
- $\sigma$ conduit à une forte probabilité de présence électronique entre les deux noyaux ;
- l'énergie de $\sigma$, qui varie avec la distance internucléaire, passe par un minimum pour 74 pm.
L'ensemble de ces résultats est synthétisé dans le « diagramme d'orbitales moléculaires » de la figure suivante.
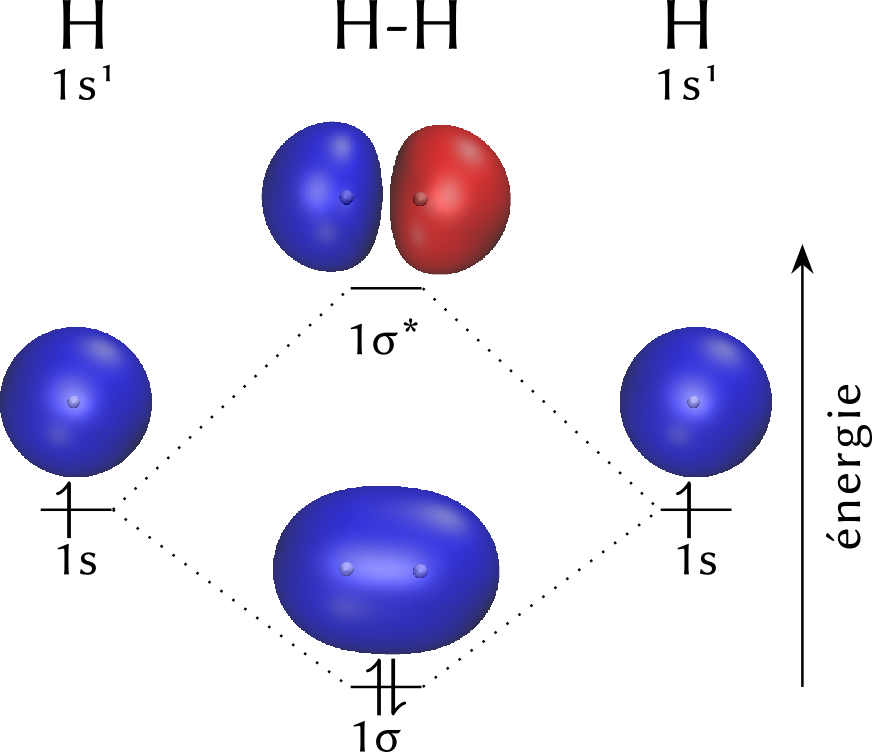
Les deux atomes isolés d'hydrogène sont représentés à gauche et à droite, avec leur configuration électronique $1s^1$. Au centre figurent les CLOA pour les deux atomes d'hydrogène rassemblés, c'est-à-dire les orbitales $\sigma$ et $\sigma^*$. Les volumes représentatifs des orbitales ont été placés au dessus des niveaux énergétiques. L'orbitale $\sigma$ ressortit de la notion de liaison covalente : les électrons des couches de valence des deux atomes sont rassemblés dans cette orbitale ; ils sont effectivement localisés entre les atomes liés et cette liaison conduit à un édifice plus sable — c'est-à-dire d'énergie inférieure — que la somme des deux atomes isolés. Cette orbitale moléculaire est dite « liante », tandis que $\sigma^*$ est qualifiée « d'antiliante ». Étant donné l'étagement énergétique des orbitales $\sigma$ et $\sigma^*$, la promotion énergétique d'un électron de l'orbitale $\sigma$ à $\sigma^*$ conduirait à une entité moins stable que la somme des deux atomes isolés : par conséquent, un apport suffisant d'énergie est susceptible de provoquer la rupture de la liaison, c'est-à-dire de l'édifice moléculaire, pour conduire aux atomes isolés. Un tel apport est possible par chauffage ou par irradiation ultraviolette ; il correspond à l'énergie de liaison $\ce{H-H}$.
La connaissance du remplissage des orbitales moléculaires permet de retrouver plusieurs caractéristiques de la molécule :
- l'ordre (ou indice) de liaison : demi-différence entre le nombre d'électrons liants et le nombre d'électrons antiliants ; l'ordre de liaison recouvre la notion de liaison simple, double ou triple des représentations de Lewis ;
- le spin $S$ total de la molécule, qui se déduit du nombre d'électron non appariés ; ici $S = \frac{1}{2} -\frac{1}{2} = 0$ puisque les deux électrons présents sont appariés et de spins opposés.
Cas du dioxygène
La méthode CLOA s'applique de manière tout à fait comparable à la molécule de dioxygène. Des restrictions s'appliquent toutefois avant d'envisager n'importe quelle combinaison linéaire :
- seules des orbitales atomiques de niveaux énergétiques comparables peuvent se combiner ;
- les orbitales doivent avoir la même caractéristique : symétrique ou antisymétrique, par rapport à leurs éléments communs : axe,
plan de symétrieCette condition est nécessaire pour que l'intégrale de recouvrement S ne soit pas nulle. .
Ainsi, dans la molécule de dioxygène et en conservant l'hypothèse de l'implication des seuls électrons de valence, les combinaisons linéaires d'orbitales atomiques pertinentes sont :
- les combinaisons entre orbitales $2s$, combinaisons qui donnent $\sigma_s$ et $\sigma_s^*$ ;
- l'axe $zz'$ étant choisi arbitrairement comme support des deux atomes, les combinaisons par un recouvrement axial des deux orbitales $2p_z$ suivant $zz'$ pour donner $\sigma_z$ et $\sigma_z^*$, cette dernière antiliante ;
- les recouvrements latéraux des deux $2p_x$, d'une part, et des deux $2p_y$, d'autre part, pour donner les orbitales moléculaires $\pi_x$, $\pi_y$, $\pi_x^*$ et $\pi_y^*$, ces deux dernières antiliantes.
Le calcul, qui sort du champ de cet exposé, montre que les énergies de ces orbitales moléculaires sont classées suivant l'ordre suivant :
$$ E_{\sigma_s} < E_{\sigma_s^*} < E_{\sigma_z} < E_{\pi_x}, E_{\pi_y} < E_{\pi_x^*}, E_{\pi_y^*} < E_{\sigma_z^*} $$La configuration électronique du dioxygène est facilement obtenue en plaçant les $6+6$ électrons de valence des deux atomes d'oxygène dans les orbitales moléculaires, comme dans la figure suivante.
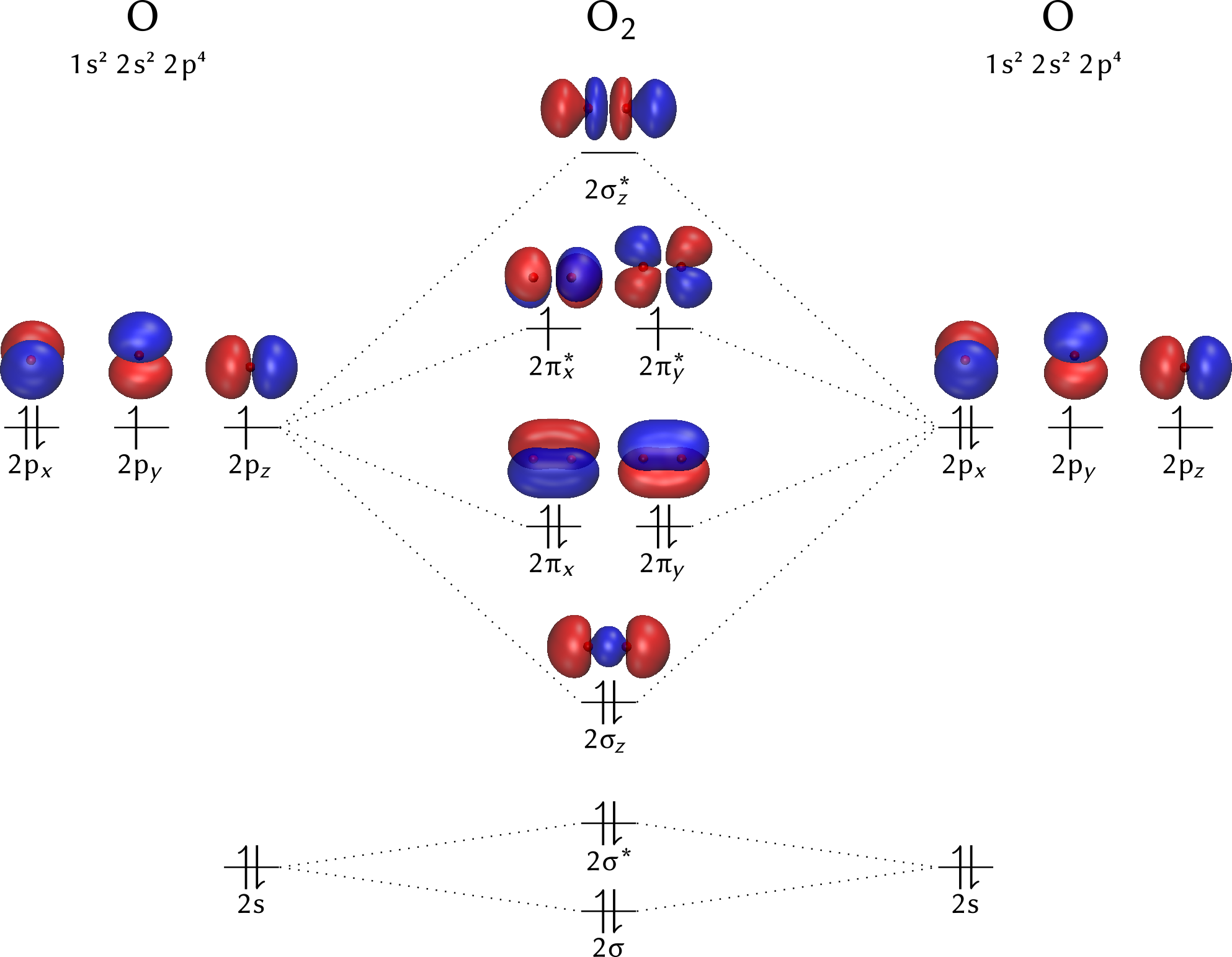
Le dioxygène possède donc deux électrons non appariés, puisque la règle de Hund impose d'affecter un électron dans l'orbitale $\pi_x^*$ et un dans l'orbitale$\pi_y^*$. L'ordre de liaison est égal à $1 -1 + 1 + 1 + 1 - \frac{1}{2} - \frac{1}{2} = 2$, tandis que le spin moléculaire vaut $S=\frac{1}{2} + \frac{1}{2}=1$, ce qui correspond au caractère paramagnétique du dioxygène, propriété observable à l'échelle macroscopique sur le dioxygène liquide. C'est notamment la valeur de ce spin qui explique les propriétés particulières du dioxygène, comme exposé dans les lignes qui suivent.
Les restrictions de spin
On définit la multiplicité $M$ d'une molécule de spin $S$ comme égale à $M =2 \times S + 1$. Ainsi une molécule dont tous les électrons sont appariés est dite « singulet » : $M = 1$ ; une molécule avec un électron célibataire est dite « doublet » $M = 2$ et une espèce à deux électrons non appariés « triplet » : $M = 3$. Le dioxygène, avec son spin $S = 1$ est une molécule triplet.
Des règles de conservation du spin ou règles de
- les réactions des triplets avec les singulets sont interdites ;
- les réactions des doublets avec les singulets sont possibles ;
- les réactions des doublets avec les triplets sont possibles ;
- les réactions des doubles entre eux sont possibles.
Par interdiction, il faut entendre : vitesse infiniment petite ou nulle et non, comme nous l'avons vu en citant quelques potentiels standards d'oxydo-réduction, impossibilité thermodynamique.
Le dioxygène dans son état électronique fondamental ne peut donc pas réagir directement avec les plupart des molécules qui nous entourent et qui sont des singulets. Force est de constater cependant que, à travers les combustions ou à travers la respiration, des oxydations sont cependant possibles. Il existe en effet des mécanismes réactionnels non directs qui permettent à l'oxygène de réagir en empruntant des voies permises, en particulier les réactions avec les doublets, c'est-à-dire les radicaux. Ces voies constituent autant de « stratégies de contournement de l'interdiction de spin ».
Stratégies de contournement de l'interdiction de spin
Rappels sur les radicaux
Dans cette partie, les radicaux ne sont pas compris comme des éléments structuraux : méthyle $\ce{-CH_{3}}$, phényle $\ce{-C_{6}H_{5}}$, etc., mais comme des espèces possédant un électron non apparié. Exemples de radicaux :
- atomes : $\quad \ce{Na} \quad 1s^2 2s^2 2p^6 \mathbf{3s^1}$ ;
- molécules : $\quad \ce{NO}$ avec 7+8 = 15 électrons au total ;
- ions monoatomiques : $\quad \ce{Fe^3+} \quad 1s^2 2s^2 2p^6 3s^2 3s^6 4s^0 \mathbf{3d^5}$ ;
Non soumis à l'interdiction de spin, puisqu'ils sont des doublets, les radicaux peuvent réagir avec le dioxygène triplet. Les produits intermédiaires obtenus, eux-mêmes radicalaires, donnent des produits finals d'oxydation et d'autres radicaux qui peuvent s'associer et terminer la réaction ou encore provoquer de nouvelles réactions radicalaires. Ainsi, les radicaux sont-ils les pivots de nombreux mécanismes de contournement de l'interdiction de spin.
Contournement radicalaire : la thermolyse par combustion
Lors d'une combustion, la température est telle qu'une partie des réactifs se décompose par rupture homolytique des liaisons $\sigma$, c'est-à-dire que l'énergie thermique permet à des électrons de passer d'une orbitale $\sigma$ à une orbitale $\sigma^*$ : les liaisons sont alors rompues pour former des radicaux. Les radicaux réagissent ensuite facilement avec le dioxygène (dont une partie a pu être rompu pour donner des atomes d'oxygène), avec les singulets présents ou encore entre eux.
Le caractère exothermique du bilan réactionnel qui permet de maintenir une température élevée : la décomposition des réactifs et la combustion peuvent se poursuivre. Sans cette température, la
Ainsi, une allumette peut enflammer une brindille mais pas une bûche : la chaleur apportée par l'allumette est suffisante pour chauffer et thermolyser la brindille, mais bien trop petite pour élever la température de la bûche. C'est pourquoi aussi un obstacle thermique peut parfois être mis en place pour empêcher la propagation d'une combustion. La flamme de la lampe des mineurs du XIXème siècle était-elle entourée d'une toile métallique : la masse du métal suffit à refroidir les gaz à son contact et à empêcher la propagation de la combustion vers l'extérieur de la lampe — c'est-à-dire la galerie de la mine — où pourrait se trouver une poche de méthane ou grisou.

Réactions en chaîne
Une des caractéristiques des réactions radicalaires est la création de chaîne : lorsqu'une espèce à nombre pair d'électrons réagit avec une autre à nombre impair, il en résulte obligatoirement au moins une espèce à nombre impair d'électrons, donc un radical, qui peut lui même réagir, etc. Un exemple souvent présenté en L1 ou L2 est celui de l'halogénation des alcanes. Il suffit qu'une réaction d'initiation fournisse les premiers radicaux pour que les étapes de propagation soient enclenchées :
- initiation $\qquad \ce{Cl–Cl -> Cl^{.} + Cl^{.}} $
- propagation $\qquad \ce{Cl^{.} + CH3–CH2-H -> HCl + CH3-CH2^{.}} $
- propagation $\qquad \ce{Cl-Cl + CH3-CH2^{.} -> Cl^{.} + CH3-CH2-Cl} $
- terminaison $\qquad \ce{Cl^{.} + CH3-CH^{.}2 -> CH3-CH2-Cl} $
- terminaison $\qquad \ce{2 Cl^{.} -> Cl–Cl} $
- terminaison $\qquad \ce{2 CH3-CH2^{.} -> CH3–CH2–CH2–CH3} $
Les étapes de propagation continuent tant qu'il y a des réactifs (ci-dessus : dichlore et éthane). En effet, les étapes de terminaison s'avèrent souvent assez lentes. Ce type de réaction se rencontre fréquemment dans les processus oxydatifs à basse température, sans thermolyse. Ainsi, le rancissement des lipides obéit-il au schéma de la figure ci dessous. S'agissant des lipides alimentaires, plusieurs mécanismes d'initiation peuvent fournir les premiers radicaux lipidiques $\ce{L^{.}}$. La figure en montre trois : le ① avec l'intervention d'un ion métallique à une degré d'oxydation élevé ; le ② par échange de radical $\ce{H^{.}}$ avec une espèce radicalaire quelconque ; le ③ par photolyse. Un des traits de l'oxydation lipidique est son caractère divergent : en effet, le produit d'oxydation primaire $\ce{LOOH}$ est un hydroperoxyde qui se décompose spontanément en deux radicaux $\ce{LO^{.}}$ et $\ce{HO^{.}}$, de sorte que le milieu réactionnel contient un nombre toujours croissant de radicaux. Ainsi, lorsque le rancissement d'une matière grasse a commencé, il est quasiment impossible de le stopper.

Catalyse métallique
Certains degrés d'oxydation des métaux de transition, en particulier au sein de complexes, autorisent des associations métal-$\ce{O2}$ sans interdiction de spin. Le complexe formé est une espèce oxydante. C'est notamment les cas dans les hèmes :

Les électrons présentent une certaine mobilité dans cette structure. Ainsi, en milieu acide, le complexe peut perdre de l'eau et donner une entité fer-oxo qui, elle aussi, est un oxydant puissant et actif. Ce nouveau complexe peut ainsi oxyder des liaisons $\ce{C-H}$ considérées comme assez inertes. C'est alors le fer (IV) de la figure suivante qui est l'agent oxydant.

Dans les êtres vivants, la grande majorité des réactions qui utilisent le dioxygène comme oxydant passent par un contournement de spin d'enzyme à cofacteur métallique. C'est la cas très notable de la respiration au niveau de la cytochrome
Oxygène singulet
Les oxydations par le dioxygène doivent emprunter les stratégies de confinement de spin en raison du caractère triplet du dioxygène. Cette caractéristique correspond à l'état fondamental et n'existe plus dans certains états excités, en particulier les états singulets. La figure suivante montre la structure électronique de deux d'entre eux, assez proches en énergie de l'état fondamental triplet.

Plusieurs processus permettent la formation de l'oxygène singulet :
- l'absorption d'un rayonnement infrarouge (1,27 µm) ou proche infrarouge (762 nm) qui apporte l'énergie pour passer de $\ce{^{3}O2}$ à $\ce{^{1}O2}$; l'interaction entre le champ électromagnétique oscillant et l'oxygène triplet est hautement improbable (toujours pour des questions de spin...) et ce processus n'est pas efficace ;
- l'absorption par un molécule $\ce{M}$ de rayonnement à une longueur d'onde inférieure à 1,27 µm pour produire une molécule excitée $\ce{M{*}}$ avec un excès d'énergie supérieur à 94,3 kJ mol-1, puis le transfert de cette énergie au dioxygène. $\ce{M}$ est appelé « photosensibilisateur ».

L'hémoglobine favorise ainsi le rancissement des lipides et, en boucherie ou en charcuterie, des coupes nettes et des tissus musculaires qui ne suintent pas sont des facteurs de stabilité à l'oxydation (sans rentrer dans l'aspect microbiologique !).
Les réactions qui impliquent le dioxygène fondent une classe particulièrement bien fournie de systèmes hors d'équilibre. Il en existe cependant d'autres comme le montre le dernier exemple abordé dans ce cours.
D'autres réactions radicalaires...
Les radicaux n'interviennent pas que dans les processus d'oxydation, même s'ils y tiennent un rôle particulier comme voie de contournement de l'interdiction de spin.
D'une manière générale, les radicaux sont des espèces très réactives. À ce titre, elles peuvent d'avérer délétère. C'est ainsi que le rayonnement ultraviolet, en apportant des photons assez énergétiques pour promouvoir des électrons liants dans des orbitales antiliantes, cassent des liaisons pour fournir, par exemple, des radicaux $\ce{C.}$ et $\ce{H.}$ Ces radicaux peuvent ensuite intervenir dans de nombreux processus :

Cette forte réactivité peut aussi être mise à profit pour exercer un effet destructeur sur des substances à éliminer. On peut citer par exemple :
- l'utilisation des ultraviolets pour la stérilisation ;
- l'élimination de la matière organique réfractaire (qui n'est pas digérée par les microorganismes) en traitement des eaux. La réaction de Fenton : $$\ce{Fe^2+ + H2O2 -> Fe^3+ + OH- + OH.} $$ donne le radical hydroxyl $\ce{OH.}$ particulièrement réactif ;
- certaines photothérapies où des porphyrines modifiées viennent se fixer sur les organes cibles : à l'irradiation, elles produisent de l'oxygène singulet, des radicaux ;
Nous illustrerons ce type d'application avec l'exemple d'un médicament anticancéreux.
Exemple de la dynémicine
Pour illustrer la complexité des phénomènes mis en jeu dans les milieux complexes comme les organismes vivants ou l'environnement, envisageons le mécanisme d'action d'un principe actif anticancéreux, la dynémicine A.
La dynémicine A est un ène-diyne, caractérisé par deux liaisons triples de part et d'autre d'une double liaison. Cet enchaînement est inclus dans une molécules de taille moyenne, dont la structure tridimensionnelle est verrouillée par une fonction époxyde
Dans l'environnement des cellules cancéreuses, le pH est souvent plus bas que dans celui des cellules saines. La dynémicine A est alors susceptible de subir le « réarrangement de Bergman » décrit dans le clip audio de la figure suivante.
Le biradical formé est extrêmement réactif : il est susceptible d'ablater un atome d'hydrogène à une espèce organique quelconque. Porté par la dynémicine, son action se concentre sur les acides nucléiques. En effet, le squelette carboné de la dynémicine A et groupes fonctionnels qu'il porte conduisent cette molécule à s'insérer dans le sillon de la double hélice de l'ADN : cette insertion est conduite par les forces de van der Waals et les liaisons hydrogène et illustrée sur la figure suivante. C'est alors sur l'ADN que la dynémicine A exerce son action délétère, conduisant à la mot de la cellule cancéreuse.
Utiliser la souris pour faire tourner la figure, en maintenant enfoncé le bouton gauche ; la molette permet de changer le zoom. Dans cette figure, un fragment d'ADN est représenté en traits fins et est entouré d'une surface verte qui correspond, en gros, à la surface de la molécule. Il n'est pas possible pour une autre molécule d'y pénétrer en raison de la gêne stérique. La molécule de dynémicine A est représentée en « balls and sticks » avec les atomes de carbone en gris, d'hydrogène en blanc, d'oxygène en rouge et d'azote en bleu selon le code couleur CPK.
Ouverture de l'époxyde en fonction du pH, insertion dans le sillon de l'ADN grâce aux liaisons faibles de van de Waals, action délétère des radicaux : l'action de la dynémicine montre combien il est nécessaire d'avoir une approche intégrative de nombreuses propriétés chimiques — et combien ces propriétés sont liées à la structure moléculaire — pour comprendre le fonctionnement de la matière qui nous entoure.
Réactivité : approche orbitalaire
Dans cette partie, nous envisagerons comment les orbitales moléculaires permettent d'analyser la réactivité.
Les interactions HOMO-LUMO : bases et acides de Lewis
En chimie organique, la description des mécanismes réactionnels passe généralement par le dessin de flèches dont il est dit « qu'elles représentent le mouvement des électrons ». Lorsqu'une flèche concerne deux entités distinctes et non un déplacement interne à une molécule, elle part d'une espèce qui :
- possède des électrons ;
- est susceptible de céder ou partager ces électrons
- présente, au moins localement, un déficit électronique ;
- est susceptible d'accepter des électrons
HOMO et LUMO
En termes d'orbitales moléculaires, une réaction base-acide se décrira comme l'interaction des électrons les « plus disponibles »,
c’est-à-dire ceux qui occupent sur une molécules l’orbitale non vide de plus haute énergie ou HOMO (Highest Occupied Molecular Orbital)
et, sur une autre molécule, l’orbitale vacante la plus facilement accessible, c'est-à-dire de plus basse énergie
ou LUMO (Lowest Unoccupied Molecular Orbital).
Si on examine par exemple la molécule de dioxygène, il y a deux HOMO qui contiennent chacune un électron : $2\pi_x^*$ et $2\pi_y^*$, tandis que
la LUMO est la $2\sigma_z^*$
Exemple de l'oxyhémoglobine
Le fer (II) de l'hémoglobine fixe le dioxygène. L'interaction entre ces deux espèces se comprend aisément en termes d'obitales moléculaires : la configuration électronique du fer (II), pris à l'état fondamental et gazeux, fait apparaître six électrons dans les cinq orbitales $3d$ : $\quad \ce{Fe^2+} \quad 1s^2 2s^2 2p^6 3s^2 3s^6 4s^0 \mathbf{3d^6}$.
Dans l'hémoglobine, les cinq orbitales $3d$ n'ont, en fait, pas la même énergie : même en l'absence de dioxygène, le fer y est déjà complexé dans un schéma octaèdrique incomplet par cinq ligands : quatre atomes d'azote qui appartiennent à l'hème de l'hémoglobine et un atome d'azote d'une histidine (résidu numéro 87) de la chaîne protéique de l'hémoglobine, comme indiqué sur la figure suivante :
Les cinq directions définies par ces cinq ligands sont orthogonales et s'organisent selon le schéma d'un complexe octaèdrique. Dans ce directions pointent les lobes de ces deux orbitales $3d$ de $\ce{Fe2+}$ : $3d_{z^2}$ et $3d_{x^2 - y^2}$, lobes qui sont orthogonaux deux à deux :
Il en résulte une levée de dégénérescence des orbitales $3d$ : les deux orbitales qui pointent vers les ligands, ligands qui portent des doubles d'électrons, sont plus hautes en énergie que les trois orbitales qui pointent dans d'autres directions de l'espace. Le schéma en énergie est donc le suivant :
Les six électrons $3d$ remplissent donc complètement des orbitales $3d_{xy}$, $3d_{xz}$ et $3d_{yz}$. Les orbitale $3d_{z^2}$ et $3d_{x^2 - y^2}$ constituent une paire de LUMO, susceptibles d'interagir avec les
électrons d'orbitales HOMO des ligands. Dans le cas du dioxygène, on voit que l'HOMO est une orbitale $2\pi^*$, à moitié remplie. L'interaction entre l'hème et
le dioxygène existe, mais elle est assez faible : on peut penser qu'il existe une lien entre ce fait et la réversibilité de l'association
dioxygène-hémoglobine. Cette réversibilité est nécessaire au transport.
L'interaction avec d'autres molécules dont l'HOMO contient deux électrons, par exemple le monoxyde de carbone ou l'anion cyanure,
est beaucoup plus forte. Ces poisons se lient de manière quasi irréversible de fer de l'hème.
Exemple d'une attaque nucléophile
De très nombreuses réactions font intervenir des attaques nucléophile. C'est par exemple la cas de l'attaque de l'ylure de la thiamine diphosphate sur le pyruvate (cette attaque est la première étape de réactions essentielles, notamment la décarboyxylation oxydative du pyruvate en acétyl-CoA). Le mécanisme commence donc par :

Cette attaque se comprend en termes d'interaction HOMO-LUMO. L'HOMO de l'ylure a l'allure suivante :
La LUMO du pyruvate a l'allure suivante :
Dans la carboxylase, le placement des réactifs permet l'interaction HOMO-LUMO
Réactifs mous et durs
L'expérience montre que le charges partielles présentes sur les sites nucléophiles et électrophiles jouent aussi un rôle dans le rapprochement des réactifs.
Finalement, suivant la structure des molécules, en particulier la présence de charges partielles et la forme des orbitales de valence, les réactions sont plutôt le résultat d'interactions électrostatiques ou plutôt l'aboutissement d'un recouvrement orbitalaire. Les concepts d'acide et base de Lewis peuvent alors être enrichis de la notion de dureté : c'est la théorie Hard Soft Base Acid ou HSBA.
Dureté
Lorsque les interactions coulombiennes dominent, la réaction est sous contrôle coulombien ; les espèces sont « dures ». Un électrophile/acide dur :
- a une LUMO de haute énergie, peu diffuse ;
- est un ion positif ou possède un site fortement chargé positivement.
Un(e) nucléophile/base dur(e) :
- a une HOMO de basse énergie, peu diffuse ;
- est un ion négatif ou possède un site fortement chargé négativement.
Mollesse
Lorsque les interactions entre orbitales dominent, la réaction est sous contrôle orbitalaire ; les espèces sont molles. Un électrophile/acide mou :
- a une LUMO de basse énergie, diffuse mais localisée sur un site ;
- peut ne pas posséder de charge ou de site fortement chargé.
Un(e) nucléophile/base mou (molle) :
- a une HOMO de haute énergie, diffuse mais localisée sur un site ;
- peut ne pas posséder de charge ou de site fortement chargé.